
| Home | Teaching | Advising | Research | Publications |
Our research primarily focuses on the development of algorithms for large-scale data analytics, with particular emphasis on network-structured data and data integration. A distinctive character of our research is that it is application-oriented, in that we aim to study application-specific problems from a computational perspective. The main application we have been focusing in the last decade has been Systems Biology, where network models are used to model interactions and associations between various components of biological systems. In recent years, we also applied what we learned from our experience with biology to such fields as energy research and intimate partner violence. An important outcome of our research is software that implements our algorithms for data analytics, which is available as open source. The following projects are among those that are currently undertaken by our group.
Phosphorylation Networks and Cellular Signaling | |
|
|
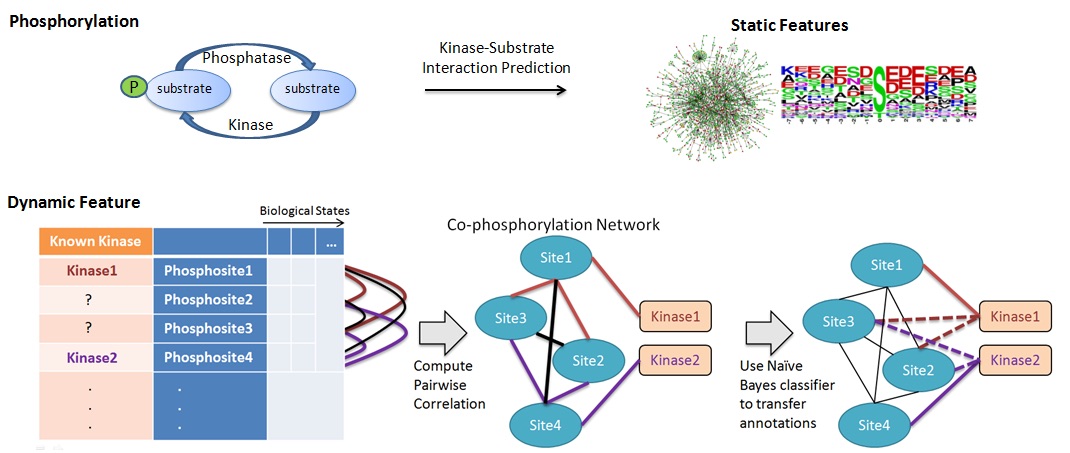
In human cells, attachment of a phosphate to a protein at certain sites can alter the activity and the function of the protein. This mechanism, known as protein phosphorylation, is often used to communicate signals within and between cells. Recent research shows that likely over 70% of human proteins can be phosphorylated. Dysregulation of protein phosphorylation is known to play an important role in many diseases, including cancer, Alzheimer's disease, Parkinson's disease, obesity and diabetes, and fatty liver disease. Indeed, many modern drugs used to treat various cancers target kinases, the enzymes that are responsible for the phosphorylation of proteins. Despite the success of the "genomic revolution" and the importance of protein phosphorylation in human biology, the knowledge on protein phosphorylation in humans is quite limited. To date, thousands of phosphorylation sites on human proteins have been discovered, but the kinases that are responsible for phosphorylating these sites could be identified for less than 5% of these sites. Recognizing the challenges associated with analyzing phospho-proteomic data, we utilize network science to extract patterns of correlation in phosphorylation levels of proteins. By organizing these patterns in "co-phosphorylation networks" and using graph-theoretic algorithms and machine learning, we extract knowledge from these networks, which are then used to develop new biological hypotheses. Besides generating basic biological knowledge such as functional annotation of phospho-proteins, kinases, and phosphatases, we also develop methods to characterize the signaling processes that are affected in cancers and Alzheimer's disease. We collaborate on this project with Mark Chance, Director of the Center for Proteomics and Bioinformatics at CWRU School of Medicine. This project is supported by National Institutes of Health grant R01-LM012980 from the National Library of Medicine. | |
Integration, Compression, and Version Control of Big Networks | |
 | |
|
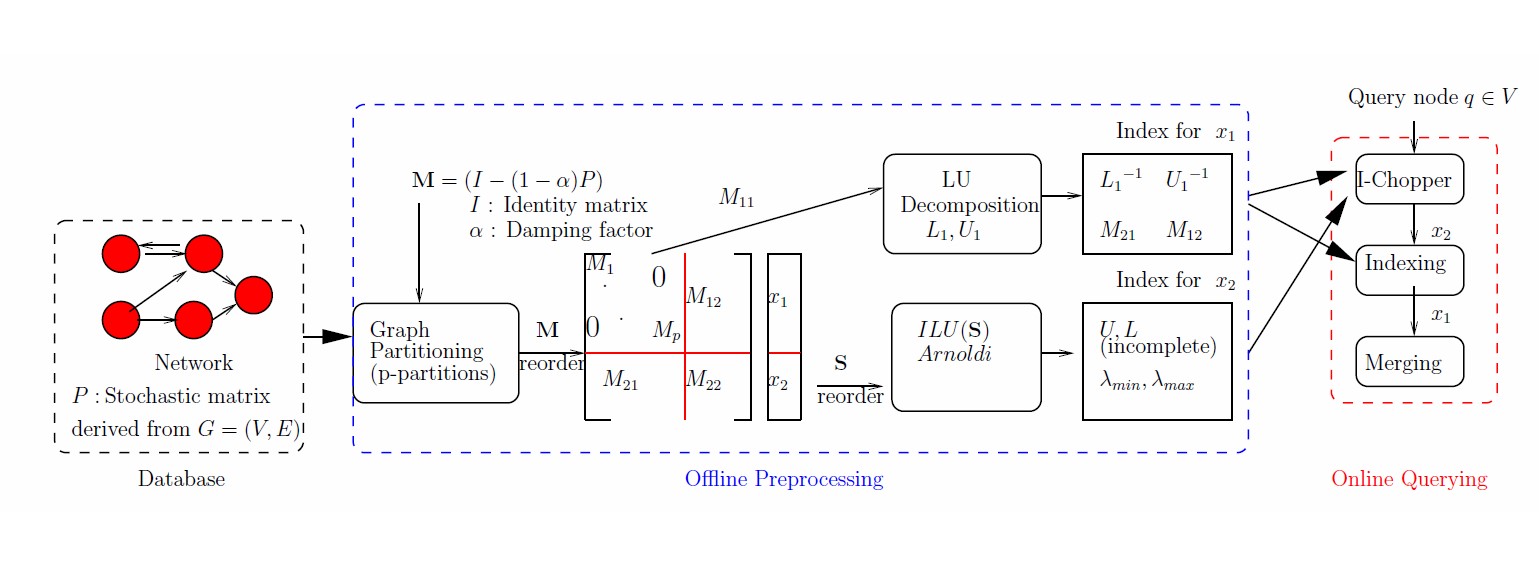
In many applications, network models are commonly used to represent interactions and higher-level
associations among various entities. Integrated analyses of these interaction and association data
has proven useful in extracting knowledge, generating novel hypotheses, and developing predictive models.
Applications include recommender systems, disease gene prioritization, network de-noising, and tracking temporal
evolution of networks.
Our research seeks to answer a number of fundamental questions that relate to efficient utilization of
large network-structured datasets: - what are (provably) optimal storage schemes for large network structured
databases? how should multiple versions of same/ related datasets be stored? how does one trade-off compression
with query efficiency? and how does one suitably abstract network data so that users can interactively interrogate
them using web-based front-ends?
To answer these questions, we develop theoretically grounded and computationally validated storage
schemes, algorithms, and software that enables efficient and effective storage, update, processing,
and querying of big and heterogeneous networks.
This project has been supported by National Institutes of Health grant
U01-CA198941 through the Big Data to Knowledge (BD2K)
program. | Characterizing the Interplay among Multiple Genetic Factors in Complex Diseases |
|
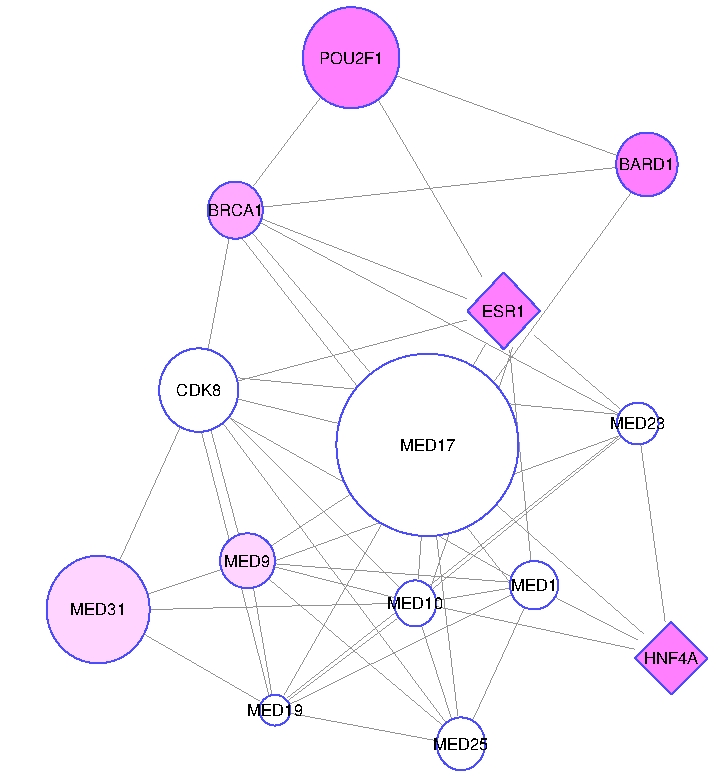
Genome Wide Association Studies (GWAS) comprehensively compare common genetic variants in affected
and control populations to identify variants that are potentially associated with complex diseases.
In recent years, GWAS successfully identified susceptible genes for many diseases. However,
researchers recognize many limitations of GWAS in characterizing the genetic bases of complex
diseases, including reduced statistical power due to small sample size, inadequacy of separate
consideration of individual variants in capturing the interplay between multiple factors,
modest success in predicting individual risk for disease, and lack of insights into the
biological and functional mechanisms that relate identified variants to the disease. We aim to
enhance GWAS by using protein-protein interaction (PPI) networks as an integrative framework to
interpret the outcome of GWAS within a functional context. PPI networks characterize the physical
and functional interactions among functional proteins; thus they are useful in understanding the
functional relationships between multiple genetic factors.
This project has been supported
by National Institutes of Health
Award R01-LM011247. |
|
Discovery of Coordinately Dysregulated Subnetworks in Complex Phenotypes | |
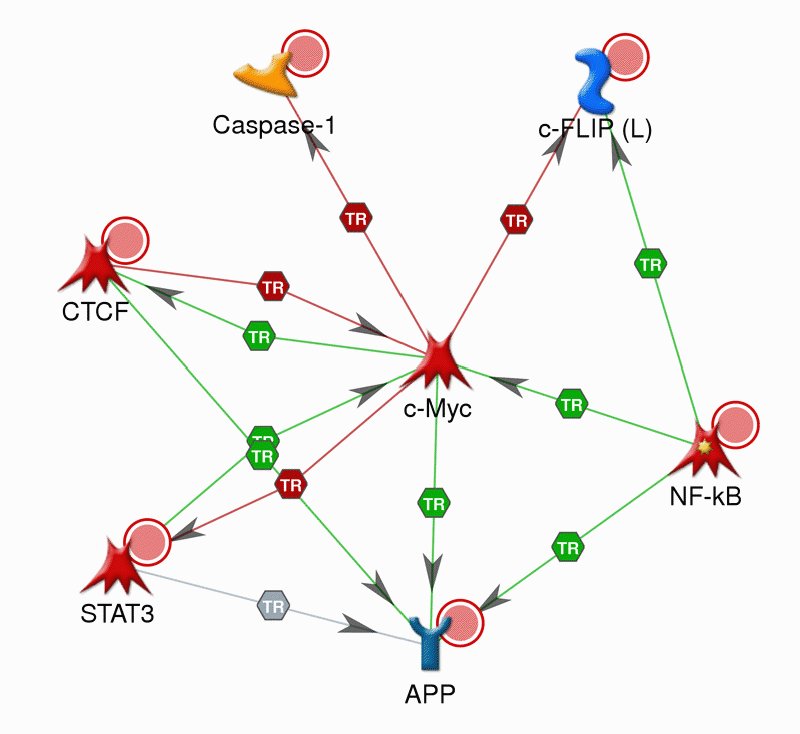 |
Cellular systems are orchestrated through combinatorial organization of thousands of biomolecules.
This complexity is reflected in the diversity of phenotypic effects, which generally present themselves as
weak signals in the expression profiles of single molecules. For this reason, researchers increasingly
focus on identification of multiple markers that together exhibit differential expression with respect
to various phenotypes. In collaboration with the research group
of Mark
Chance, we focus on human colorectal cancer and develop abstractions and algorithms
to define coordinate dysregulation of multiple genes within network context and identify such network
patterns with a view to establishing markers for prognosis of cancer and targets for theurapetic
intervention. For this purpose, our algorithms integrate genomic, transcriptiomic, proteomic, and interactomic
data. This project is supported in part by NSF CAREER Award
CCF-0953195. |